- Get link
- X
- Other Apps
This is a short story about the simplified mechanism behind a potentially fatal arrhythmia -- torsades de pointes (which means "twisting of the peaks" in French). In this post, we expect the readers know the Nernst equation and the origin of resting membrane potential and action potential. For those who don't know that yet, related videos could be easily found in websites like Khan Academy. (https://www.khanacademy.org/science/health-and-medicine/nervous-system-and-sensory-infor/neuron-membrane-potentials-topic/v/neuron-resting-potential-description)








Fig.1. Electrocardiography(ECG) of torsades de pointes, showing the ventricular arrhythmia with constantly rotating cardiac axis. A heart with an electrical activity like this is not able to contract coordinately to produce meaningful cardiac output. (Figure source: https://lifeinthefastlane.com/ecg-library/tdp/)
Before we start, we need to know how electrical activities normally transmit through the heart. Normally, electrical impulse transmit in a sequential way with the aids of the conduction system. The activated tissue are temporarily block from further action potential because of the refractory period. During the absolute refractory period, the voltage-sensitive sodium(Na) channels, the main channels bringing up the potential changes during action potential, on the membrane of cardiomyocytes are already open and thus cannot be turned on further. While during the relative refractory period, the voltage-sensitive Na channels are slowly recovered from inactive state and thus it is still possible to trigger an action potential by large enough voltage changes. The refractory period ensures sequential contraction of cardiac tissues that produces meaningful cardiac output.

Fig.2. The transmission of electrical impulse through the heart. (Figure source: Guyton & Hall Textbook of Medical Physiology 12ed)
However, in some cases, the depolarization and even the activation of threshold could occur during the early phase of relative refractory period, which is called early afterdepolarization(EAD). To understand how EAD is generated, we need to recall the electrophysiology of cardiomyocytes.

Fig.3. Early afterdepolarization and delayed afterdepolarization(not stated in the main text). The early afterdepolarization is caused by the abnormal reactivation of L-type Ca channel, which will be explained in the main text. (figure source: Harrison's Principle of Internal Medicine, 17th edition.)
The action potential is a triggered positive feedback when the membrane potential is raised beyond a threshold: the depolarization turns on more voltage-sensitive channels, and these channels bring up currents that further depolarize the membrane. This positive feedback phenomenon originates from antagonizing current pairs. Take the threshold near the resting membrane potential as an example, the inward currents of inward rectifier K channels and the outward currents of Na channels form an antagonizing current pair. The summation current-voltage curve of these 2 antagonizing currents has 3 fixed points. The fixed point near -70mV is an unstable fixed point, and if the membrane potential is brought beyond this point, it will trigger a positive feedback that brings the membrane potential near +50mV.

Fig.4. The antagonizing current pair made of the inward rectifier K currents (the red curve) and outward Na currents (the blue curve). The summation current-voltage curve (the green curve) has 3 fixed points. The fixed point near -70mV is an unstable fixed point, and if the membrane potential is brought beyond this point, it will trigger a positive feedback that brings the membrane potential near +50mV. This is the basis of physiological threshold in ventricular cardiomyocytes.
However, this is not the only pair of antagonizing currents possible in cardiomyocytes. The action potential of ventricular cardiomyocyte could be divided into 4 phases. In phase 4, inward rectifier (which means the ion channel preferentially allow inward current) K channels sets the resting membrane potential at about -85mV. In phase 0, the Na channels open and bring the membrane potential to about +20mV. The transient outward K channels briefly open and bring the membrane potential to about 0mV, making up phase 1. In phase 2 and phase 3, L-type Ca channels and delayed rectifier K channels form another antagonizing current pair that keeps the membrane potential near 0mV for a prolonged time period and brings up the plateau shape of the action potential. However, due to the intracellular calcium dependent inactivation of L-type Ca channel, the Ca current gradually fades out and the K current takes over, bringing the membrane potential back to -85mV.
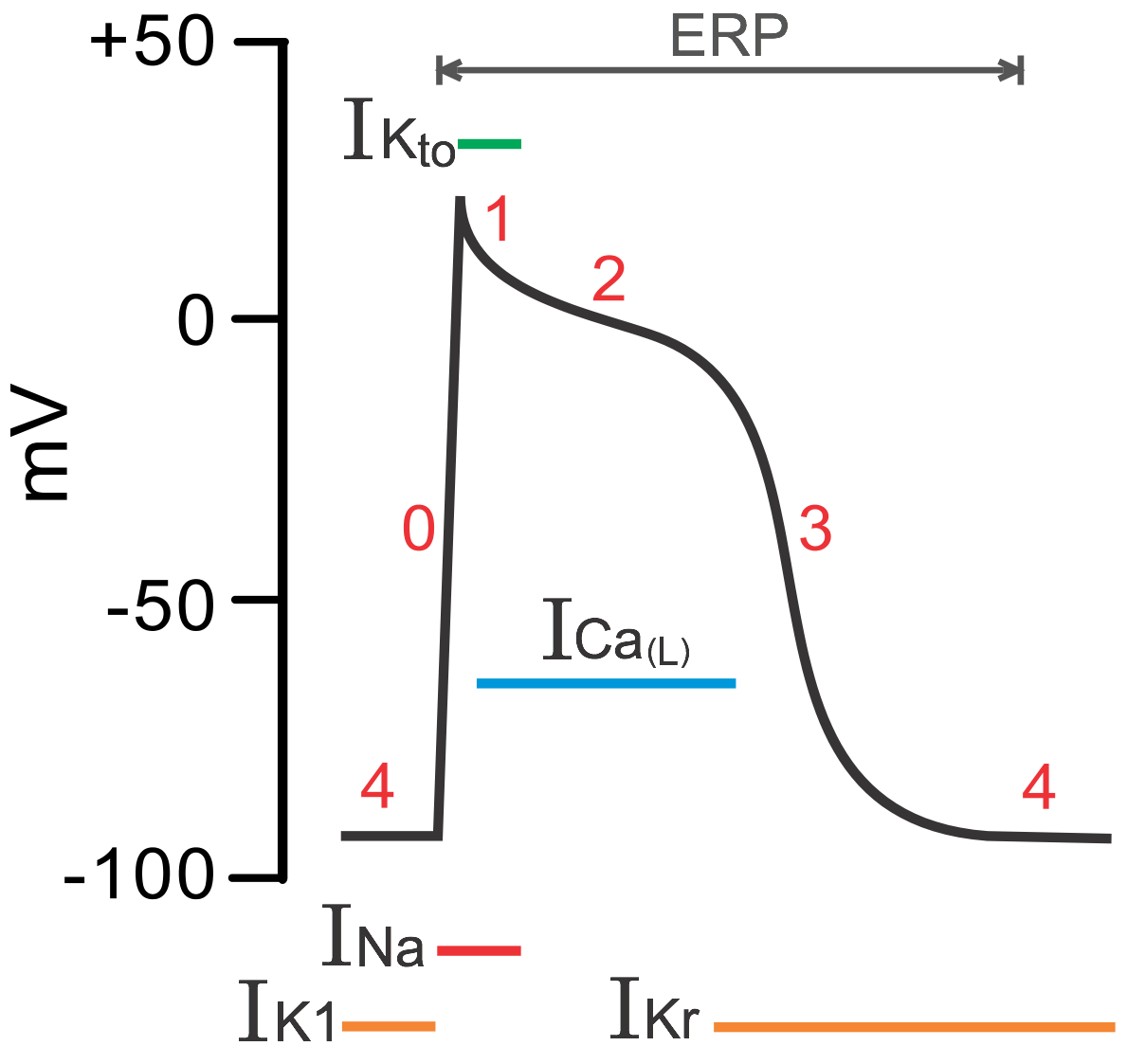
Fig. 5. Four phases of the action potential of cardiomyocytes. (Figure source: https://www.cvphysiology.com/Arrhythmias/A006.htm)
This could be displayed in a more figurative way like Fig. 6. The antagonizing current pair formed by delayed rectifier K channels and L-type Ca channel. The Ca current is suppressed by intracellular Ca dependent inactivation, so the Ca current is not large enough to bring up 3 fixed points in the summation current-voltage curve. No pathological threshold is allowed, and there is therefore no EAD.

Fig.6. The antagonizing current pair formed by delayed rectifier K channels and L-type Ca channel. The Ca current is suppressed by intracellular Ca dependent inactivation, so the Ca current is not large enough to bring up 3 fixed points in the summation current-voltage curve. No pathological threshold is allowed, and there is therefore no EAD.
However, if the action potential is prolonged, the Na-Ca exchanger (NCX) would clear up the intracellular calcium. The intracellular Ca dependent inhibition of L-type Ca channel is lifted up and the L-type Ca channel is ready to fire again. As shown in Fig. 7, the summation current-voltage curve again has 3 fixed points, and the unstable fixed point is the pathological threshold. The early afterdepolarization could activate itself and adjacent cardiomyocytes in a disorganized way and cause torsades de pointes.

Fig.7. Prolonged action potential allows the Na-Ca exchanger to clear up intracellular calcium. The L-type Ca channel is able to open again and the summation current-voltage curve has 3 fixed points. This pathological threshold is the basis of early afterdepolarization.
It is known that torsades de pointes is induced by increased sympathetic tone and some electrolyte imbalance such as hypokalemia, hypocalcemia, hypomagnesemia and certain drugs. Let's see how the mechanisms mentioned above work in the real world. The sympathetic tone activates β1 receptor on cardiomyocytes, which ultimately causes the phosphorylation of L-type calcium channel, calcium pump on smooth ER and the myosin light chain and activate them. Importantly, the phosphorylated L-type calcium channel is not inactivated by intracellular calcium. The effect of sympathetic tone is therefore two-fold. It not only abolishes the intracellular calcium dependent inactivation but also promote the clearing of intracellular calcium, which favor the pathological threshold.
Hypokalemia inactivates the rapidly activating delayed rectifier through unknown mechanism and overall decreases the total potassium current, which prolongs the action potential. The prolonged action potential allows the clearing of intracellular calcium and reactivates the L-type calcium channel, so the pathological threshold is allowed.
Hypocalcemia decreases the driving force of calcium current and causes less calcium accumulated in cytoplasm. Because less L-type calcium channel is inactivated, the action potential is prolonged and the pathological threshold is allowed.
Magnesium is a natural antagonist of calcium channel through unknown mechanism. Its role in suppressing trigger activity is two-fold as well. First, it blocks N-type calcium channel at the axon terminal and blocks the release of norepinephrine. Second, it blocks L-type calcium channel of cardiomyocyte no matter it’s phosphorylated or not. Therefore, magnesium blocks the sympathetic effect on cardiomyocyte and also shortens the action potential, while hypomagnesemia allows the generation of pathological threshold through the opposite.
It’s important to recognize that several predisposing factors could occur together. For example, after acute myocardial infarction, the increased sympathetic tone activates β1 receptor on cardiomyocytes, abolishes the intracellular calcium dependent inactivation, and promotes intracellular calcium clearing. It could also activate β3 receptor on adipocytes and promote free fatty acid release, which decreases serum magnesium level through saponification. All these factors favor the pathological threshold.
By understanding the mechanism deeply, we could easily know why we should use magnesium sulfate to treat TdP. Most importantly, always remember to check the electrolytes for they have profound effects on the electrical activities of cardiomyocytes.
Comments
Post a Comment